Whole-genome sequencing and analysis of Plasmodium falciparum isolates from China-Myanmar border area
Hai-Mo Shen1,2, Shen-Bo Chen1,2, Yan-Bing Cui1,2, Bin Xu1,2, Kokouvi Kassegne1,2, Eniola Michael Abe1,2, Yue Wang3,4* and Jun-Hu Chen1,2*
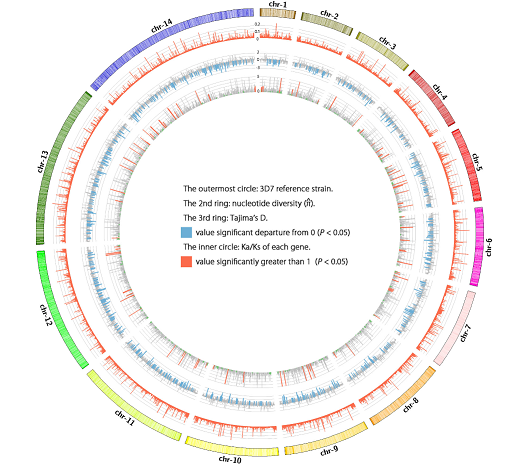
China has made progress in malaria control and aims to eliminate malaria nationwide, but implementing effective interventions along the border regions remain a huge task. The Plasmodium falciparum cases imported from Southeast Asia has frequently reported especially in the China-Myanmar border (CMB) area. Though, information is scant on P. falciparum genetic variability in this area. This study reported P. falciparum isolates genome sequence of six clinical isolates in the CMB area. Furthermore, we estimated the nucleotide diversity, Watterson’s estimator and Tajima’s D value for the whole genome mutation rate in slide window. Our data were aligned onto 96.05–98.61% of the reference 3D7 genome in high fold coverages. Principal component analysis result showed that P. falciparum clustered generally according to their geographic origin. A total of 91 genes were identified as positive selection with Ka/Ks ratio significantly higher than 1, and most of them were multigene families encoding variant surface antigens (VSAs) such as var, rif and stevor. The enrichment of the positive selection on VSA genes implied that the environment complexity subjected CMB’s P. falciparum to more pressure for survival. Our research suggests that greater genetic diversity in CMB area and the positive selection signals in VSA genes, which allow P. falciparum to fit the host immune system well and aggravate the difficulty of treatment. Meanwhile, results obtained from this study will provide the fundamental basis for P. falciparum population genomic research in CMB area.
Infect Dis Poverty. 2018 Nov 13;7(1):118. doi: 10.1186/s40249-018-0493-5.